
Phenotypic variation for germination and seedling traits under PEG-induced drought stress
The various phenotypic traits related to seeds and seedlings, their abbreviations, and formulas to calculate the corresponding derived traits are presented in Table 1. A large amount of phenotypic variation was observed for all the traits in the mini-core collection both under control (Supplementary Table 1) and chemically induced drought (20% PEG) (Supplementary Table 2). PEG treatment caused significant negative impact on both the seed traits evaluated in this study -germination percent and germination rate (Fig. 1). There were nine accessions which exhibited 100% germination in response to PEG, while 10 accessions showed less than 50% germination under PEG conditions.
Boxplot analysis of variation of minicore population for seed traits under control and drought stress (PEG 20%). (A) Germination percentage; (B) Germination pace.
All the shoot traits evaluated (Area, Diameter, Length, Volume, Weight) were significantly lower in the PEG treatment compared to the control group (Fig. 2). Similarly, all the root traits were significantly lower in the PEG treatment compared to the control group, except for root diameter. Broad sense heritability and genetic variance estimates from ANOVA are summarized in Table 2. The wide range of phenotypic variation in conjunction with the high heritability of the evaluated phenotypes lends well for the genetic dissection of these traits by GWAS.
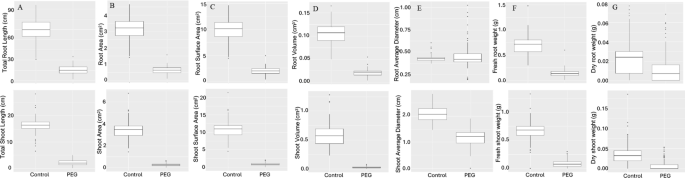
Boxplot analysis of variation in minicore population for seedling traits under control and drought stress (20% PEG). (A) Length; (B) Area; (C) Surface area; (D) Volume; (E) Diameter; (F) Fresh weight; (G) Dry weight. Top panel is associated with roots and bottom panel is for shoots.
Phenotypic variation for short-term drought stress during heading
The measurements of root weight, shoot weight, and seed yield per plant were collected in greenhouse conditions for both irrigated plants and plants subjected to a five day drought during heading (Supplementary Table 3). Significant variation in the heading time has been observed for this population. The earliest heading was 71 days after planting, while the latest was 138 days. The median heading time was 91 days. With an arbitrary cutoff ≤ 30% yield loss after short-term water stress, five lines were identified in the mini-core collection that can be considered as potential drought tolerant lines.
Correlation analysis
Correlation matrix was generated to evaluate the relationships between the various phenotypic traits evaluated in this study (Fig. 3). Significant correlations between traits in the control (Fig. 3A) and PEG/drought treatment group (Fig. 3B) were found. Closely related traits such as fresh weight, dry weight, length, area, and volume were consistently positively correlated with each other for both the roots and shoots. However, root diameter was negatively correlated with root length both in the control (-0.40) and in PEG treatment (-0.58). Shoot fresh weight and root fresh weight showed a significant positive correlation in control (0.64) and in the PEG treatment (0.27). Germination rate and root diameter showed a negative correlation in control (-0.2) and in the PEG treatment (-0.28). Germination percentage and germination rate were positively correlated (0.39) only under PEG treatment. Germination percentage showed a positive correlation with root: shoot fresh weight under control (0.22) and a negative correlation under PEG (-0.37).

Correlation analysis of seed germination, seedling traits, and mature plant traits under control and drought stress. (A) Control; (B) PEG 20% or 5-days of drought during heading.
For Drought Tolerance Index (DTI), most correlations were positive owing to a reduction commonly seen across phenotypes in response to the PEG treatment (Fig. 4A). However, positive correlations were observed (0.38) between germination percentage and germination rate, fresh weight of shoots and germination percentage (0.25), germination pace (0.23) and fresh weight of roots (r = 0.41). The negative correlation (-0.38) between total fresh weight and shoot fresh weight is indicative of the strong treatment effect imposed by PEG (Fig. 4B).

Drought tolerance index of seed germination, seedling traits and mature plant traits. (A) Box plot analysis. (B) Correlation analysis.
The reduction correlation matrix (Supplementary Fig. 2) displayed similar patterns as the DTI matrix, with most correlations being positive. In this index, root and shoot fresh weights were again found to be positively correlated (r = 0.57). The negative correlation between seed yield and germination rate (-0.25) was unexpected and warrants further investigation.
Under control conditions, root: shoot weight ratios was positively correlated with root weight (0.33) and negatively correlated with shoot weight (-0.38) (Fig. 3A). Mature shoot weight was negatively correlated with seed yield (-0.48) under drought stress conditions (Fig. 3B). Interestingly, under drought stress, shoot weight was negatively correlated with root length, area, and volume (-0.27), whereas, seed yield under irrigated conditions were positively correlated with total root length (0.17), root area (0.19) and root volume (0.20). The most interesting observation was a positive correlation (0.53) between the DTI seed yield and root dry weight (Fig. 4B). Of the nine lines that exhibited 100% germination in PEG treatment, three of them maintained large root lengths in seedling stage and furthermore, two of these lines had no significant impact on seed yields in response to drought stress during heading.
Genome wide associations
Filtering of genotypic data included using a minor allele frequency (MAF) ≥ 0.05, and minimum call rate of 80%. Following filtering, 155 accessions and 39,789 SNPs remained, of which 36,421 markers were identified as informative. SNP markers were evenly dispersed throughout the seven chromosomes, and more densely covered genomic regions with higher frequencies of recombination, such as telomeres (Supplementary Fig. 3).
GWAS analysis of the 155 spring barley accessions was conducted to identify QTLs responsible for the genetic basis of germination and seedling development. The FarmCPU modeled GWAS analysis detected 224 significant marker-trait associations with a –log10 p-value ≥ 4.65, which was set by an FDR of 0.01 and was spread across all seven barley chromosomes (Fig. 5). The physical location of significant SNPs was used to identify candidate genes on the reference genome MorexV3 pseudomolecules assembly. SNPs which were within linkage disequilibrium (LD) ±0.5 MB window, identified via LD decay, were co-located, and reported as a single marker association (Fig. 6). Further, only those significant SNPs accounting for 5% or more of the phenotypic variance explained (PVE) were considered for further analysis. This stringent filtering criteria led to 87 high quality SNPs associations (22 for control, 51 for PEG treatment, 12 for greenhouse drought treatment) across the seven barley chromosomes (Supplementary Table 4).

Manhattan plots of genome-wide association studies in barley minicore population using FarmCPU model. (A) Measured traits (B) Traits evaluated using the scanner (C) Drought Tolerance Index traits (D) Reduction traits. Significant SNPs are above the blue horizontal line (Y-axis: –log10 p-value ≥ 4.65).

Pairwise linkage disequilibrium of identified SNPs in GWAS analysis separated by treatment group. Panel A = control group; panel B = PEG-induced treatment group; panel C = 5-day short term drought during heading stage. LD coefficients are reported as r2 over genetic distance in base pairs.
Of the 22 SNPs in the control group, four SNPs were associated with two or more traits and 11 SNPs were unique to one particular trait (Supplementary Table 4). Eight SNPs were located on chromosome 2, two on chromosomes 4 and 5, and one each on chromosomes 1, 3, and 6.
For the 51 SNPs identified in association with the PEG treatment group, seven SNPs were associated with two or more traits while 31 SNPs were unique to one particular trait. In descending order, eight SNPs were localized on chromosome 5, chromosomes 2 and 7 each had seven SNPs, chromosome 6 had six SNP associations, chromosomes 1 and 3 had five SNPs, and chromosome 4 had the least with two SNPs.
Of the 12 SNPs in the greenhouse study, three SNPs were associated with two traits and six SNPs were unique to one particular trait. Five of the SNPs were on chromosome 2 and one SNP was associated each on chromosomes 4, 5, 6, and 7.
Germination percentage
In total, five SNPs showed significant associations (–log10 p-value ≥ 4.65) with germination percentage trait on four different chromosomes (Table 3, Supplementary Table 4). The SNP on chromosome 6 (JHI-Hv50k-2016-369413) identified in control, was the most significant association accounting for 88.6% of PVE. The two significant SNPs under induced drought were on chromosomes 5 (JHI-Hv50k-2016-134977) and 2 (JHI-Hv50k-2016-310413), accounting for 17.5% (p-value = 1.72 e− 5) and 9.3% (p-value = 1.8e− 5), respectively, for the variation in germination percentage. One SNP (JHI-Hv50k-2016-420812) on chromosome 6 was associated with DTI of germination percentage, explaining 27.1% of the variation (p-value = 1.52e− 5) and with reduction of germination percentage trait explaining 26.5% phenotypic variance (p-value = 1.77e− 5).
Germination rate
For germination rate, all three SNPs identified were observed in response to PEG treatment. Two SNPs (JHI-Hv50k-2016-353238 and JHI-Hv50k-2016-352386) in proximity on chromosome 5 were associated with DTI, explaining 10.9% and 5.5% of the variation, respectively. One SNP (JHI-Hv50k-2016-206096) on chromosome 3 associated with the reduction of germination rate, accounted for 9.1% of the variation in this trait (p-value = 4.74e− 6) under PEG-induced drought.
Length
Twenty significant SNPs were associated with various root and shoot length traits. These associations were made across all barley chromosomes, except chromosome 1. Of the 20 SNPs, nine were associations with the control group and 11 were in response to PEG-induced drought.
Root length
For root length, three SNPs were identified, of which, two SNPs were associated under control conditions and one SNP under induced drought. The SNP (JHI-Hv50k-2016-90004) on chromosome 2 explained 10.8% of the variation (p-value = 1.83e− 5) and the second SNP (JHI-Hv50k-2016-306304) on chromosome 5 accounted for 12% of the variation (p-value = 2.19e− 5). The SNP (JHI-Hv50k-2016-438327) on chromosome 7 explaining 27.5% of the variation in root length (p-value = 2e− 5) was identified in the PEG treated group.
Shoot length
The one SNP (JHI-Hv50k-2016-103434) on chromosome 2 found in relation to total shoot length in the control group explained 7% of the variation in this trait (p-value = 2.51e− 8). For the shoot length trait, two SNPs were identified under control conditions, and both were located on chromosome 2. One of the SNPs (JHI-Hv50k-2016-90029) explained 7.8% of the phenotypic variance (p-value = 2.27e− 8), while the second SNP (JHI-Hv50k-2016-93576) accounted for 10.7% of the variation in shoot length, albeit at a lower significance (p-value = 6.26e− 6).
Root: shoot length
Among secondary traits associated with length, 14 SNPs were associated with the root-to-shoot ratios. For this trait’s associations, two SNPs (JHI-Hv50k-2016-438327 and JHI-Hv50k-2016-143568) identified from the PEG treatment were on chromosomes 7 and 2. These two SNPs explain 14.2% (p-value = 5.59e− 6) and 12.5% (p-value = 6.42e− 6) of the phenotypic variance associated with root: shoot total length, respectively. Furthermore, these two SNPs were associated with more than one trait. The next three SNPs were associated with root: shoot ratios for the total length, one SNP was correlated with the control group and two in the PEG treated group. The single SNP identified in the control group (JHI-Hv50k-2016-94418) was located on chromosome 2 and accounted for 11.6% PVE (p-value = 2.19e− 5). Six SNPs related to length were associated with average root-to-shoot measurements. Interestingly, the three SNPs identified in control group and three SNPs in the PEG treatment group were all found on different chromosomes. The three control group SNPs were located on chromosome 2 (JHI-Hv50k-2016-103250), 4 (SCRI_RS_165031), and 5 (JHI-Hv50k-2016-352376). While the simulated drought SNPs were identified on chromosome 3 (JHI-Hv50k-2016-210371), 6 (JHI-Hv50k-2016-374365), and 7 (JHI-Hv50k-2016-438327) .
DTI-Length
Five SNPs were associated with different DTI length traits. Three SNPs were found in relation to the DTI of total length root-to-shoot ratios on chromosomes 2, 3, and 7. The most significant SNP of these three (JHI-Hv50k-2016-438327) explained 8.7% PVE (p-value = 1.9e− 6), followed by (JHI-Hv50k-2016-143568) 7% PVE (p-value = 9.79e− 6), and the final SNP on chromosome 3 (JHI-Hv50k-2016-158478) explained 6.7% of the variation (p-value = 1.21e− 5). The other two DTI associated SNPs were for average root length. Both SNPs were localized on chromosome 3, the SNP (JHI-Hv50k-2016-212430) explained 14.5% (p-value = 5.32e− 8) and the second SNP (JHI-Hv50k-2016-210371) accounted for 6% of the variation (p-value = 1.23e− 8).
Area
Six significant SNP associations were identified for the area traits of roots and shoots. As expected, the genomic regions associated with the area and surface area traits of roots and shoots overlapped.
Root area
Two SNPs were found in association with the root area trait. The SNP (JHI-Hv50k-2016-90004) associated with the control group’s root area and surface area localized on chromosome 2 and explained 23.9% phenotypic variance (p-value = 6.41e− 6). The second associated SNP (JHI-Hv50k-2016-461178) was identified in the PEG treated group, and was located on chromosome 7 explaining 28.3% phenotypic variance (p-value = 1.93e− 5).
Shoot area
For the shoot area trait, two significant SNP associations were determined. The first SNP in association returned the highest significance level for the control group (p-value = 5.32e− 9). This highly significant SNP (JHI-Hv50k-2016-103200) was on chromosome 2 and explained 5.5% phenotypic variance for shoot area and surface area. The second SNP (JHI-Hv50k-2016-155905) found in association with the control group shoot area and surface area was located on chromosome 3 with a PVE of 11.9% (p-value = 5.64e− 7).
Root: shoot area
Two SNPs related to root: shoot area ratios were identified. The first SNP (JHI-Hv50k-2016-103250) on chromosome 2 was associated with the control group for the area and surface area of R: S. This first SNP accounted for 12.4% PVE (p-value = 1.01e− 7). The next SNP (JHI-Hv50k-2016-143568) on chromosome 2 associated with the DTI of biomass area and surface area in the R: S ratio. The second SNP accounted for 31.5% (p-value = 1.01e− 5) of the variance and was also associated with the trait DTI root: shoot length.
Diameter
Six SNPs were identified in relation to the average diameter of seedlings. Five associations occurred within the induced drought group. Two SNPs (JHI-Hv50k-2016-225661 and BOPA2_12_30531) associated with average root diameter were located on chromosomes 4 and 5 explaining 12% (p-value = 3.79e− 6) and 18.5% phenotypic variance (p-value = 2.28e− 5), respectively. The third SNP (JHI-Hv50k-2016-223231) associated with the average diameter of shoots on chromosome 3 accounted for 25.7% of the variation in this trait (p-value = 5.58e− 8). The two SNPs associated with root: shoot ratio diameter was localized on chromosome 1, one was related to average measurement and the other in reduction of diameter ratios. The SNP associated with average R: S diameter (JHI-Hv50k-2016-49703) explained 12.3% PVE (p-value = 1.26e− 8) and the other SNP associated with reduction in R: S diameter (JHI-Hv50k-2016-14614) accounted for 16.9% PVE (p-value = 4.55e− 6).
Volume
Two SNPs were associated with root volume, one each in control and PEG treated group. The SNP (JHI-Hv50k-2016-90004) on chromosome 2 explained 36.8% phenotypic variance (p-value = 1.81e− 5) and was identified in the control group. The second SNP (JHI-Hv50k-2016-459904) located on chromosome 7 (8.6% PVE; p-value = 9.66e− 7) was identified in the PEG-treated group.
Weight
Most of the SNP associations were identified in relation to root and shoot weight traits. Of the 28 SNPs, 16 SNP associations were related to fresh weights and 12 to dry weights.
Fresh weight
For fresh weights, one SNP (JHI-Hv50k-2016-404363) was associated with root weight, two SNPs (JHI-Hv50k-2016-49477 and JHI-Hv50k-2016-82132) for shoot weight, two SNPs (BOPA2_12_20831 and BOPA2_12_11449) for R: S weight, three SNPs (JHI-Hv50k-2016-70142, JHI-Hv50k-2016-407667, and JHI-Hv50k-2016-291104) for the DTI of root weight, two SNPs (BOPA2_12_20326 and JHI-Hv50k-2016-36022) for the DTI shoot weight, three SNPs (SCRI_RS_133042, BOPA2_12_20831, and BOPA2_12_11449) for the DTI of R: S, one SNP (JHI-Hv50k-2016-71911) for the reduction of root weight, and two SNPs (JHI-Hv50k-2016-462613 and JHI-Hv50k-2016-439099) for reduction of shoot weight. All SNPs that were not DTI or reduction calculations were found in the PEG-treatment group except for one SNP (JHI-Hv50k-2016-82132), which was in the control group and associated with shoot weight. Five SNPs were localized on chromosome 2, two SNPs each on chromosomes 1,4,5,6, and 7. The most significant (p-value = 9.42e− 7) SNP associated with reduction of shoot weight was located on chromosome 7 and explained 5.5% of the phenotypic variance associated with this trait.
Dry weight
SNPs associated with dry weights were distributed on six chromosomes. This included two SNPs (JHI-Hv50k-2016-45487 and SCRI_RS_184126) for root weight in the control group, one SNP (SCRI_RS_182407) for shoot weight in PEG treated group, a SNP (JHI-Hv50k-2016-297664) for the DTI of R: S weight, four SNPs (JHI-Hv50k-2016-33403, JHI-Hv50k-2016-445646, JHI-Hv50k-2016-121548, JHI-Hv50k-2016-432785) for reduction of root weight, and one SNP (JHI-Hv50k-2016-438195) for the reduction of shoot dry weight.
Short-term drought in greenhouse
For the five-day terminal drought stress experiments, 23 significant SNP associations were identified that spanned all seven chromosomes. Twelve of these SNPs accounted for more than 5% of the variation in the traits. In this group of 12, seven SNPs were on chromosome 2, two SNPs on chromosome 4, and one SNP each on chromosomes 5, 6, and 7 (Table 3). Three SNPs (JHI-Hv50k-2016-70895, JHI-Hv50k-2016-250342, BOPA2_12_30531) associated with seed yield reduction in response to drought were found on chromosomes 2, 4, and 5, and explained 25%, 10.4% and 11.8%, respectively. Two SNPs on chromosome 2 (JHI-Hv50k-2016-68926 and JHI-Hv50k-2016-122309) were associated with the DTI of root: shoot weight. Two additional SNPs (JHI-Hv50k-2016-100142 and JHI-Hv50k-2016-68926) were associated with a reduction in root: shoot weight. Three SNPs were identified in the control plants (JHI-Hv50k-2016-68167 on chromosome 2, JHI-Hv50k-2016-250342 on chromosome 4, JHI-Hv50k-2016-489288 on chromosome 7) that were associated with shoot weight, seed yield, and root weight, respectively. The final two SNPs (JHI-Hv50k-2016-122309 and JHI-Hv50k-2016-377967) were associated with root: shoot weight ratios and root dry weight. Both SNPs were identified in drought-stressed plants and accounted for 28% and 30% of the variation in these traits, respectively.
In silico gene expression of candidate genes
Genes located within the ± 0.5 Mbp window of LD of each significant marker were extracted identifying a total of 1446 genes. To reduce the number of genes for further consideration we concentrated on SNPs that were located inside the predicted genes and within their putative promoter regions (2 Kb) (Table 3). This caveat drastically reduced the potential candidate genes to 54. Most of the 54 genes showed differential expression (2-fold) in the 16 different tissue types that were reported (Supplementary Table 5). Fifteen genes were differentially expressed in the seedling stage comparisons involving nine different barley genotypes. In the comparison of three tissue types- radicle, plumule, and scutellum across seven different timepoints during seed germination, more than 25 of the potential candidate genes were differentially expressed. This finding further corroborates the significant marker-trait associations identified in this GWAS. The most interesting differences for the candidate genes were identified in the transcriptome analysis involving drought treatments. This involved three different studies wherein comparisons were made between drought tolerant and drought sensitive lines. Twenty genes were identified as being differentially expressed between the drought stressed and control samples (Supplementary Table 5).



